A review on nonsteroidal anti-inflammatory drugs (NSAIDS)
S. K. Mandal
Faculty of Pharmaceutical Chemistry, Dr. B. C. Roy College of Pharmacy
& AHS, Durgapur- 713206, India.
Corresponding author:
Mr. S. K. Mandal, Assistant Professor, Faculty of Pharmaceutical Chemistry,
Dr. B. C. Roy College of Pharmacy & AHS, Durgapur- 713206, India.
E-mail: gotosudip@rediffmail.com, Tel.: +918670192100
.
ABSTRACT
NSAIDs constitute an important class of drugs with therapeutic applications
that have spanned several centuries. Treatment of inflammatory conditions
such as rheumatoid arthritis (RA) and osteoarthritis (OA) starting from the
classic drug aspirin to the recent rise and fall of selective COX-2
inhibitors has provided an enthralling evolution. This review traces the
origins of NSAIDs, their mechanism of action at the molecular level such as
cyclooxygenase (COX) inhibition, inflammatory Process, biosynthesis of
prostanoids, COX isoforms, their structure, function and comparison,
development of selective COX-2 inhibitors, adverse gastrointestinal effects
of NSAIDs. The presence of COX-3 is discussed. A little
history of the market withdrawal of selective COX-2 inhibitors is
explained. The last section describes briefly some of the
recent advances toward developing effective anti-inflammatory agents such
as nitric oxide donor NO-NSAIDs, dual COX/LOX inhibitors and Hydrogen
sulfide (H2S) Containing NSAIDs. In spite of the tremendous
advances in the last decade, the design and development of a safe,
effective and economical therapy for treating inflammatory conditions still
presents a major challenge.
Keywords:
Nonsteroidal, Inflammation, Prostaglandin Cyclooxygenase, Lipoxygenases.
INTRODUCTION
The fascinating ability to treat fever and inflammation dates back about
3500 (400 B.C.) years ago to a time when the Greek physician Hippocrates
prescribed an extract from willow bark and leaves. Later in the 17th
century, the active ingredient of willow bark salicin was identified in
Europe. The Kolbe company in Germany started mass producing salicylic acid
in 1860. Acetylsalicyclic acid 1 (aspirin) the more
palatable form of salicyclic acid was introduced into the market by Bayer
in 1899. However, the mechanism of action of anti-inflammatory and
analgesic agents such as aspirin and indomethacin 2
remained elusive until the early 1960’s. This all changed in the seventies,
when John Vane discovered the mechanism of action of aspirin and other
nonsteroidal anti-inflammatory drugs (NSAIDs) thereby increasing our
ability to develop novel anti-inflammatory therapies. The success of NSAIDs
in treating various inflammatory conditions such as rheumatoid arthritis
(RA) and osteoarthritis (OA) validated inhibition of the enzyme
prostaglandin H synthase (PGHS) or cyclooxygenase (COX) as a highly
suitable target in anti-inflammatory therapies. However, the
gastrointestinal (GI) toxicities associated with widespread NSAID use
proved to be a major drawback during long term therapy2.
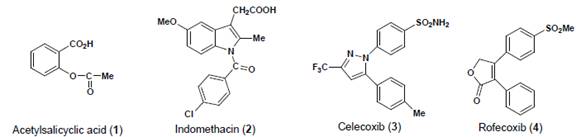
Figure 1:
Some representative examples of NSAIDs
In the early 90’s, Needleman, Simmons and Herschman’s group reported the
presence of an inducible isoform of the enzyme COX later identified as
COX-2. This discovery led to the hypothesis that anti-inflammatory
prostaglandins (PGs) were produced through constitutive expression of
COX-1, whereas the proinflammatory PGs were produced via induction of the
COX-2 isoform. In 1999, G.D. Searle and Pfizer (now Pfizer Inc) launched
the first selective COX-2 inhibitor celecoxib 3
(Celebrex®). This was followed by the launch of Merck’s selective COX-2
inhibitor rofecoxib 4 (Vioxx®). In spite of this initial
success after the launch of selective COX-2 inhibitors, concerns were
raised regarding their adverse cardiovascular demonstrated that selective
COX-2 inhibitors may tip the natural balance between prothrombotic
thromboxane A2 (TxA2) and antithrombotic prostacyclin (PGI2) potentially
increasing the possibility of a thrombotic cardiovascular event 3,4. In April of 2005, the US FDA advisory committee
overwhelmingly concluded that coxibs increase the risk of cardiovascular
events and recommended the suspension of Pfizer’s Bextra® (valdecoxib).
Celecoxib was allowed to remain in the market place, but with a black box
warning indicating a risk of adverse cardiovascular events5.
Health Canada recently decided to withdraw Novartis Pharmaceuticals
selective COX- 2 inhibitor lumiracoxib (Prexige®) due to concern regarding
its liver toxicity2. Recently, the American Heart Association
issued a statement advising prescribing clinicians pertaining to the use of
NSAIDs6.
INFLAMMATORY PROCESS
Inflammation is a biochemical and cellular response that occurs in all
vascularized tissue whose health and vitality is threatened by either an
internal or an external source. Most of the essential components of the
inflammatory response can be found in the blood, and most of the early
mediators (facilitators) of inflammation function to increase the movement
of plasma and infection fighting blood cells from the capillary bed into or
around the injured tissue. Collectively known as exudate, usually a clear
serous fluid, these substances defend the host against infection and
facilitate tissue repair and healing7. The superficial hallmarks
of inflammation have been described since antiquity. They are: • Redness
(rubor) • Heat (calor) • Pain (dolor) • Swelling (tumor) • Loss of function
(functio laesa).
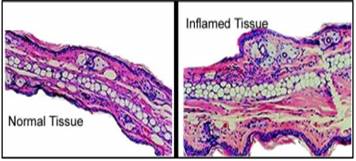
Figure 2:
Histological sections through a normal and an inflamed retina. (age-related
macular degeneration)7
On a microscopic level, three characteristic changes in the
microcirculation occur near the site of tissue injury. i) Increased blood
flow to the area. ii) Increased vascular permeability which allows leakage
of plasma into the damaged area. iii) An increased number of white blood
cells immigrating through vessel walls to the site of injury. Histological
sections through a normal and an inflamed retina shown in (Figure 2).
BIOSYNTHESIS OF PROSTANOIDS
PGs and thromboxane A2 (TXA2), collectively termed
prostanoids, are formed when arachidonic acid (AA), a 20-carbon unsaturated
fatty acid, is released from the plasma membrane by phospholipases (PLA 2) and metabolized by the sequential actions of PGG/H synthase
or by cyclooxygenase (COX) and their respective synthases.There are 4
principal bioactive PGs generated in vivo: prostaglandin E2 (PGE 2), prostacyclin (PGI2), prostaglandin D2
(PGD2), and prostaglandin F2α (PGF2α). PG
production (Figure 3) depends on the activity of PGG/H synthases,
colloquially known as COXs , bifunctional enzymes that contain both COX and
peroxidase activity and that exist as distinct isoforms referred to as
COX-1 and COX-28-9.
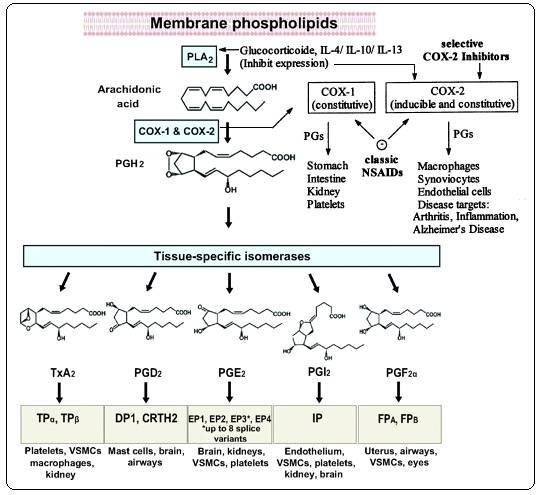
Figure 3:
Biosynthesis of Prostanoids and site of action of anti-inflammatory drugs 8-9
PGH2 is produced by both COX isoforms, and it is the common
substrate for a series of specific isomerase and synthase enzymes that
produce PGE2, PGI2, PGD2, PGF2α
, and TXA2. COX-1 couples preferentially, but not exclusively,
with thromboxane synthase, PGF synthase, and the cytosol (c) PGE synthase
(PGES) isozymes.COX-2 prefers prostaglandin I synthase (PGIS) and the
microsomal (m) PGES isozymes, both of which are often coinduced along with
COX-2 by cytokines and tumor promoters. Prostanoids exert their actions on
other cells through various G-protein coupled receptors.
CYCLOOXYGENASES
The COX isoforms are heme containing enzymes that exhibit distinct
expression profiles and roles in several physiological processes. The first
crystal structure of ovine COX-1 complexed with the NSAID flurbiprofen was
reported in 1994. The structures of human and murineCOX-2 are virtually
super imposable on ovine COX-1. Comparison of the COX-1 and COX-2 isoforms
given in Table1.The COX isoforms are homodimers, with each monomer
comprised of three structural domains; a N-terminal epidermal
growth factor (EGF) domain, a membrane binding domain (MBD) and a large
Cterminal catalytic domain (Figure 4). The COX catalytic reaction occurs in
a hydrophobic channel in the core of the enzyme while the peroxidase site
is located in the heme containing region near the protein surface. The MBD
is made up of four alpha helices with helix D merging into the catalytic
domain. These helices surround an opening through which fatty acid
substrates and NSAIDs are believed to enter the COX active site. N
-glycosylation of the COX isoforms is required for enzyme folding and
activity 2.
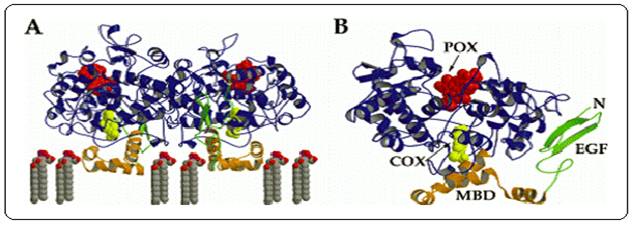
Figure 4:
A.
diagram of the ovine COX-1 homodimer with flurbiprofen bound within the COX
active site. B. diagram of ovine COX-1 monomer with
flurbiprofen bound indicating the locations of the COX and peroxidase (POX)
active sites and the EGF and MBD domains. Flurbiprofen is represented as a
yellow space filling model2.
Table 1:
Comparison of the COX-1 and COX-2 isoforms 10
|
Parameter
|
COX-1
|
COX-2
|
|
Regulation
|
Usually Constitutive
|
Inducible
|
|
Range of Induced Gene Expression
|
2 to 4-fold
|
10 to 80-fold
|
|
Rate of Gene Activation
|
24 hours
|
0.5 to 4 hours
|
|
Effect of Glucocorticosteroids
|
Little or None
|
Inhibits Expression
|
|
Relative Size of Active Site
|
Smaller
|
Larger
|
|
Rate of Arachidonic Acid Consumption
|
34 nmol/min/mg
|
39 nmol/min/mg
|
|
Effect of aspirin on COX activity
|
Inhibited
|
Not Affected
|
PRESENCE OF COX-3?
A new twist was added to the COX story in 2002 with the discovery of a
third isoform COX-3 by Simmons and coworkers. Their study in dogs showed
that COX-3 was present as an alternative splice variant of COX-1 11-12. The Simmons group showed that indeed COX-3 was the target
of acetaminophen. However, the initial excitement surrounding the discovery
of COX-3 as a potential drug target received a reality check when it was
discovered that one can not generalize the presence of canine COX-3 to
humans. It is now known that COX-3 encodes proteins with completely
different amino acid sequences than COX-1 or COX-2 in rodents and humans
and moreover lacks COX activity. This negates its role in causing pain and
fever. Therefore, the clinical relevance of COX-3 as a drug target is
questionable. However the final jury on this question is not out yet 13-14.
MECHANISM OF ACTION OF NSAIDS
A simplified explanation of the effect of inhibitors of the COX enzymes is
as follows. The carboxyl moiety of acidic NSAIDs interacts with Arg120 in
both COX isoforms, via hydrogen bonding or electrostatic interactions. The
remaining ligand-protein interaction is hydrophobic. Most NSAIDs act
reversibly, mainly by excluding arachidonate, but aspirin binds to and
acetylates the serine at position 530 causing irreversible inactivation of
the enzymes.
The crucial differences between the two COX enzymes (Figure 5) are at
position 523: here COX-1 has bulky isoleucine while COX-2 has valine
smaller molecule that leaves a gap, which gives access to a side pocket. It
is this side pocket that is believed to be the binding site for COX-2
selective agents, which in general have a rigid side-extension that can
reach across the channel and interact with the pocket. This aspect of their
structure appears to be the basis of their selectivity for COX-2: they may
be too bulky to fit into the COX-1 channel16.
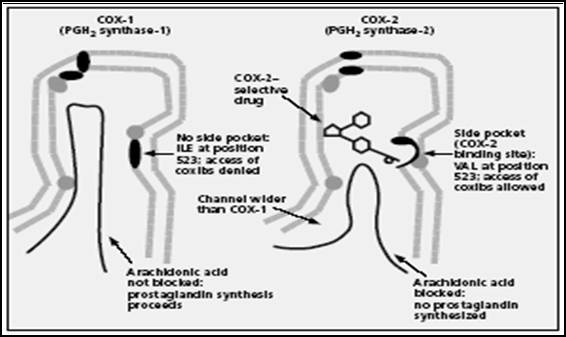
Figure 5:
Structure of the COX-1 and COX-2 enzymes. Schematic showing active site
similarities and differences16.(ILE = isoleucine; VAL = valine.)
MANAGEMENT OF NSAIDS RELATED GASTROINTESTINAL TOXICITY
Current hypotheses for roles of COX-1 and COX-2 in the pathogenic mechanism
of NSAID-induced gastric damage. The motility hypothesis suggests that
gastric motility plays an important role in NSAID damage. NSAIDs induce
vagal-dependent gastric hypermotility via inhibition in COX-1-mediated
prostaglandin (PG) production and CNS actions. Subsequent microvascular
disturbances lead sequentially to neutrophil-endothelial interaction and
oxyradical production. Inhibition of COX-1 leads to up-regulation of COX-2
expression17. PG production mediated by COX-2, which may
suppress the neutrophil-endothelial interaction, is also decreased by COX-2
selective or nonselective NSAIDs (Figure 6). The
neutrophil-endothelial interaction plays a major role in the neutrophil
hypothesis, which suggests that NSAIDs activate the neutrophil through
alteration of arachidonic acid metabolites (eg, PGs), enhancing
neutrophil-endothelial cell adhesion.
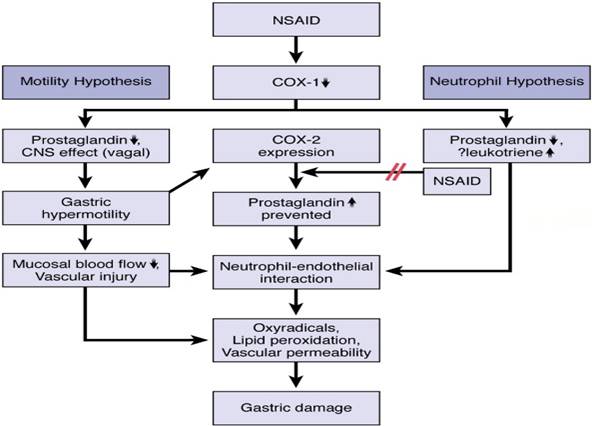
Figure 6: Roles of COX-1 and COX-2 in the pathogenic mechanism of
NSAID-induced gastric damage17
Comparisons of gastric damage and cyclo-oxygenase (COX) selectivity of
nonsteroidal anti-inflammatory drugs (NSAIDs). The left-hand side of the
(Figure 7) represents the adjusted relative risk for bleeding and
perforation of the upper gastrointestinal tract. Values for
anti-inflammatory doses of NSAIDs are shown18. The right-hand
side of the figure represents log COX-2 : COX-1 activity ratios (IC 50 values; μmol/L) for these NSAIDs
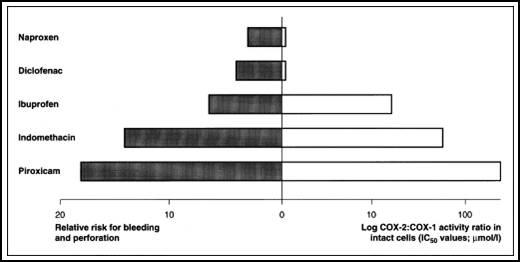
Figue 7:
Comparisons of gastric damage and COX selectivity NSAIDs18.
CONVERSION OF NONSELECTIVE COX INHIBITORS TO COX-2-SELECTIVE INHIBITORS
15, 19
Modifying well known NSAIDs into selective COX-2 inhibitors represents an
interesting strategy. Indomethacin, zomepirac, aspirin and flurbiprofen
have been successfully elaborated into selective COX-2 inhibitors (Figure
8). However, the methodology utilized in NSAID modification does not follow
a general scheme. Classic NSAIDs such as indomethacin possess both COX-1
and COX-2 inhibiting activity. Various attempts have been made to shift the
enzyme selectivity of indomethacin from COX-1 to COX-2 while keeping the
potency on the same level and reducing the unwanted side-effects at the
same time.
In principle, the strategy consisted of introducing larger substituents to
fit into the active site volume of COX-2 (L-748780). Introducing a larger
trichlorobenzoyl analogue instead of the chlorobenzoyl analogue optimized
COX-2 selectivity. A similar strategy was used for the modification of
zomepirac, basically a COX-1 selective drug. The desired COX-2 selectivity
was achieved by replacing the acetic acid group by other moieties such as
the pyridazinone ring or an N-acyl aminosulfonyl phenyl group to
yield RS-57067 and RS-1048934, respectively. In contrast
exchanging the carboxylate moiety of the aspirin with alkyl sulfide
functionalities afford specific COX-2 inhibitors.
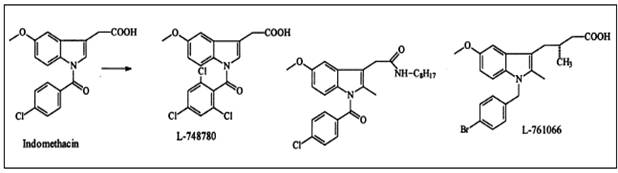
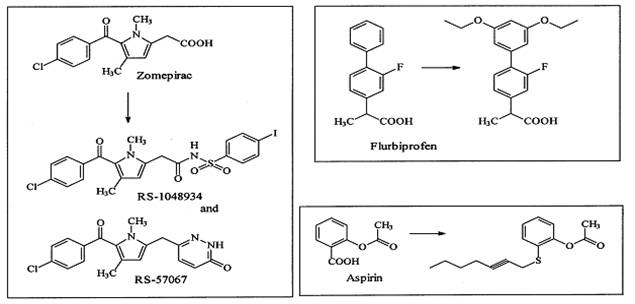
Figure 8:
Conversion of nonselective COX inhibitors to COX-2-selective inhibitors 15
DUAL COX AND LIPOXYGENASE (LOX) INHIBITORS
It is well known that arachidonic acid (AA) primarily undergoes
biotransformation to proinflammatory and anti-inflammatory PGs via COX
mediated isoform catalysis. Lipoxygenases (LOXs), which belong to a class
of non-heme ironcontaining enzymes, catalyze dioxygen incorporation into
AA, to form hydroperoxide products. For example, AA metabolism catalyzed by
5-LOX affords proinflammatory leukotrienes (LTs) that may play a role in
cardiovascular diseases since they are potent vasoconstrictors . In
addition, other LOX mediated metabolites such as cysteinyl-LTs are known to
cause GI mucosal damage.
ML-3000 (licofelone)(1)exhibits dual COX and 5-LOX
inhibitory activities. Licofelone exhibited effective anti-inflammatory
activities with reduced GI toxicities in animal models. Preliminary data in
humans have shown that licofelone could be an alternative to NSAIDs in
treating OA. In this regard, compound-(2) possesses the
pyrazole ring system present in the selective COX-2 inhibitor celecoxib in
conjunction with the 5-LOX pharmacophore present in the marketed drug
ZD-2138. This dual inhibitor exhibited excellent COX-2
inhibitory potency and selectivity along with potent 5-LOX inhibition 20. The propynone (3) exhibited selective COX-2
inhibition and 5-LOX inhibition along with in vivo anti-inflammatory
activity in animal models. In another study compound-(4)
evaluated in this study exhibited dual COX/LOX inhibition. Related studies
targeted to the design of novel COX/LOX inhibitors as effective
anti-inflammatory agents with reduced side effects have been reported. The
molecular structures of few potent dual inhibitors are given in (Figure 9)
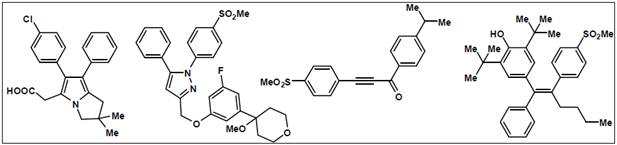 (1)
(2)
(3)
(4)
(1)
(2)
(3)
(4)
Figure 9:
Molecular structures of dual inhibitors2, 20
licofelone (2-[6-(4-chlorophenyl)-2, 2-dimethyl-7-phenyl-2,
3-dihydro-1H-pyrrolizin-5-yl] acetic acid) have been found to be
significantly effective in Phase III clinical trials conducted on patients
of osteoarthritis21. A series of novel acrylic acid derivatives (I, II) were designed and synthesized bearing at the 3
position thienyl, furfuryl and 3,5-ditert-butyl-4-hydroxyphenyl
substituents and tested as potential dual lipoxygenase/cyclooxygenase-1
(LOX/COX-1) inhibitors and as anti-inflammatory agents22.
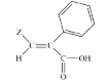
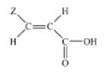
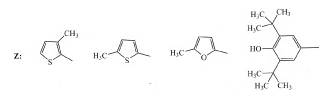
(I) (II)
NITRIC OXIDE (NO) CONTAINING NSAIDS
The first reports describing NO-NSAIDs began to appear in the literature
during the 1990’s. NO-NSAIDs were investigated with the objective of
abolishing the GI toxicity associated with traditional NSAID therapy since
NO was known to protect the GI mucosa. These studies showed that hybrid
NO-NSAIDs exhibited efficient anti-inflammatory activities without causing
GI side effects. The recent adverse cardiovascular events associated with
selective COX-2 inhibitor therapy has provided a strong stimulus for the
development of NO-NSAIDs since NO exhibits beneficial cardiovascular
effects such as vasodilation, and inhibition of platelet aggregation 23.
In this regard, a novel class of pyrazole analogs developed as selective
COX-2 inhibitors containing nitrate groups as hybrid-NO donors. Compound (a) (Figure 10) exhibited potent COX-2 inhibition and
selectivity in conjunction with good GI tolerance (safety). An alternate
approach also described wherein the central furanone ring system of
rofecoxib was replaced by a furoxan ring. This concept was based on the
observation that a furoxan ring system can act as a NO-donor. Therefore,
3,4-diphenylfuroxans were designed for evaluation as hybrid
COX-inhibitor/NO donors. Within this class of compound, the furoxan (d) exhibited selective COX-2 inhibition in conjunction
with NO-donor properties. NO-NSAIDs such as aspirin, naproxen, and
diclofenac have been investigated the most. In the majority of these
studies, organic nitrates or nitrosothiols have been employed as the
NO-donor group2. However, long term treatment with organic
nitrates can cause “nitrate tolerance” leading to lack of GI and
cardiovascular benefits. To counter this problem NO-NSAIDs containing novel
diazonium-diolate groups developed that have the potential to theoretically
release two molecules of NO with half-lives that correlate well with their
pharmacological durations of action . The aspirin (b) and
ibuprofen (c) hybrid NO-donors exhibited effective
anti-inflammatory activity with reduced or no GI toxicities. Compounds (e) and (f) exhibited COX-2 selectivity
as well as vasodilator properties.
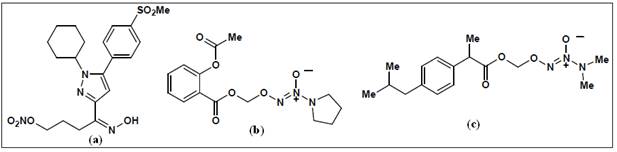
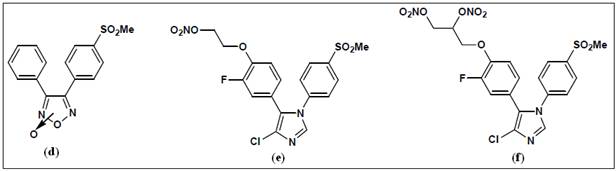
Figure 10:
Examples of hybrid NO-NSAIDs2, 23.
HYDROGEN SULFIDE (H2S) CONTAINING NSAIDS
Hydrogen sulfide, H2S, is a colorless gas with a strong odor
that until recently was only considered to be a toxic environmental
pollutant with little or no physiological significance. However, the past
few years have demonstrated its role in many biological systems and it is
becoming increasingly clear that H2S is likely to join nitric
oxide (NO) and carbon monoxide (CO) as a major player in mammalian biology.
An overview of the chemistry and biology of H2S and have
summarized the chemistry and biological activity of some synthetic H2S-donating compounds have provided (III). The synthetic H 2S donating NSAIDs of aspirin, sulindac, diclofenac,
indomethacin, and ibuprofen have been reviewed in detail24. The
newly reported NOSH-aspirin (IV) that releases both NO and H2S
has also been discussed.
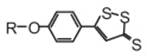

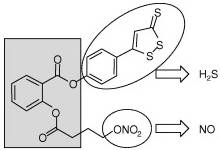
(III)
(IV)
NOSH–aspirin
A study said that a molecule, which releases hydrogen sulphide - the gas
that gives rotten eggs their characteristic smell- have an
anti-inflammatory effect. The team hopes that using H2S donating molecules
to control H2S delivery in the body could pave the way for the development
of novel approaches to the treatment of inflammatory. He discovered that
when H2S is delivered in a slow and sustained manner, a potent
anti-inflammatory effect is produced. The cell signalling molecules that
drive inflammation, such as TNFa, IL-1, IL-6 and prostaglandins, were
reduced while levels of the body''s own anti-inflammatory molecules (i. e.
IL-10) were increased25.
When hydrolyzed, H2S-releasing NSAIDs produce the parent NSAID
and the H2S-releasing moiety from which H2S is
released (Figure 11).The NSAID component inhibits COX-1 and COX-2 resulting
in compromised mucosal defense mechanisms, which may lead to ulcers. NSAIDs
can reduce renal perfusion, which can lead to increases in blood pressure
(BP) leading to cardiovascular (CV) damage. The released H2S
counteracts many of the detrimental effects of NSAIDs. These protective
effects appear to be mediated through activation of KATP
channels. H2S enhances the mucosal defense mechanisms; causes
vasodilation thus reducing BP leading to cardioprotective effects. Both the
NSAID and H2S have anti-inflammatory effects, the former through
inhibition of COX and latter through inhibition of nuclear transcription
factor κB (NF-κB).
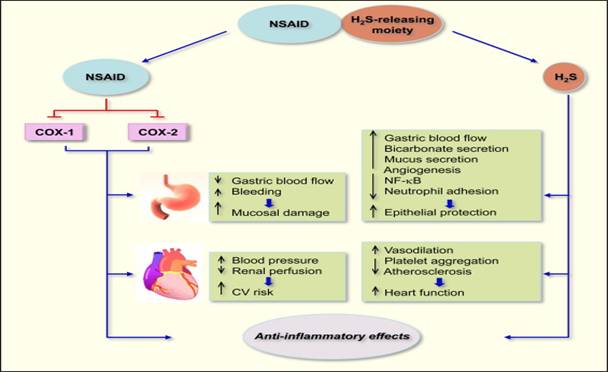
Figure 11:
Mechanisms of action of H2S-releasing NSAIDs24.
FUTURE PROSPECTS
Various epidemiological and laboratory studies have indicated that NSAIDs
may be able to reduce the risk of cancer (colorectal cancer in particular),
Angiogenesis and Alzheimer's disease due to their inhibitory activity on
COXs, especially COX-222.
A constitutive overexpression of COX-2 seems to be important in colon
carcinogenesis. In cultured human colonic fibroblasts it was shown that
growth factors such as hepatocyte growth factor are involved in the
progression of tumors. COX-2 inhibitors are now assumed to inhibit
COX-2-mediated PG synthesis which is responsible for hepatocyte growth
factor expression.
It is hypothesized that tumor-derived growth factor promotes angiogenesis
by inducing the production of COX-2-derived PGE2. PGs are known
to be pro-angiogenic molecules and contribute to tumor growth by inducing
the newly formed blood vessels (neoangiogenesis) that sustain tumor cell
viability and growth.
Recent results indicate an important role of COX-2 in the central nervous
system (CNS). COX-2 expression is markedly induced in CNS neurons by
excitatory stimuli such as ischemia and seizures so that a role of COX-2
derived PGs in certain forms of neurodegeneration can be assumed. The fact
that COX-2 mRNA is elevated in areas related to memory (hippocampus,
cortex) and that the amount of COX-2 correlates with the deposition of
beta-amyloid protein represents a possible therapeutic benefit and a
hopeful new strategy in the prevention or treatment of Alzheimer's Disease
(AD). It has also been shown that celecoxib maximally inhibits COX-2 in the
CNS at anti-inflammatory doses.
CONCLUSIONS
NSAIDs represent an important class of compounds. The rapid discovery of
selective COX-2 inhibitors can be attributed to the rational drug design
approach. However, the gastrointestinal adverse effects of traditional
NSAIDs and the cardiovascular adverse effects associated with selective
COX-2 inhibitors highlights the pitfalls that may be encountered in the
drug discovery paradigm. NO-NSAIDs, H2S-releasing NSAIDs and
dual COX/LOX inhibitors represent novel approaches directed toward the
development of effective anti-inflammatory therapy. In spite of the
unprecedented advances in drug discovery, developing a safe, effective and
economical therapy for treating inflammatory conditions still presents a
major challenge2.
ACKNOWLEDGMENT
The author is grateful to the principal Dr. Subrata Chakraborty and the
management, especially to Mr. Dulal Mitra, President, BCREC Society for
their cooperation.
REFERENCES
[1]. Vane. J.R. The fight against rheumatism: from willow bark to COX-1
sparing drugs. J Physiol Pharmacol. 2000, 51, 573-586.
[ 2.] Rao, P.N.P.; Knaus E.E. Evolution of Nonsteroidal Anti-Inflammatory
Drugs (NSAIDs): Cyclooxygenase (COX) Inhibition and Beyond. J Pharm Pharmaceut Sci. 2008, 11,
81-110.
[3]. Bresalier, R.S.; Sandler, R.S.; Quan, H.; Bolognese, J.A.; Oxenius,
B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A. et al.
Cardiovascular events associated with rofecoxib in a colorectal adenoma
chemoprevention trial. N Engl J Med. 2005, 352, 1092-1102.
[4]. Solomon, S.D.; McMurray, J.J.; Pfeffer, M.A.; Wittes, J.; Fowler, R.;
Finn, P.; Anderson, W.F.; Zauber, A.; Hawk E.; Bertagnolli. M.
Cardiovascular risk associated with celecoxib in a clinical trial for
colorectal adenoma prevention. N Engl J Med. 2005, 352, 1071-1080,
.
[5]. Dogné, J.M.; Supuran, C.T.; Pratico. D. Adverse cardiovascular effects
of the coxibs. J.Med. Chem. 2005, 48, 2251-2257.
[6]. Antman, E.M.; Bennett, J.S.; Daugherty, A.; Furberg, C.; Roberts, H.;
Taubert. K.A. Use of nonsteroidal antiinflammatory drugs: an update for
clinicians: a scientific statement from the american heart association.Circulation, 2007, 115, 1634-1642 .
[7]
http://prosono.ieasysite.com/patho_chapter_inflammatory_response.pdf
(Visited
January, 2013)
[8]
Ricciotti
, E.;
FitzGerald
, G.A. Prostaglandins and Inflammation Arteriosclerosis.Thrombosis and Vascular Biology. 2011, 31, 986-1000.
[9] Dannhardt, G.; Kiefer, W. Cyclooxygenase inhibitors – current status
and future prospects. Eur. J. Med. Chem. 2001, 36,109–126.
[10] Smith, W.L.; DeWitt, D.L.; Garavito, R. M. Cyclooxygenases:
structural, cellular, and molecular biology. Ann Rev Biochem, 2000, 69,145- 182,
[11] Chandrasekharan, N.V.; Dai, H.; Roos, K. L.; Evanson, N. K.; Tomsik,
J.; Elton, T. S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited
by acetaminophen and other analgesic/antipyretic drugs: cloning, structure,
and expression. Proc. Natl. Acad. Sci. 2002, 99,13926-13931.
[12] Chandrasekharan, N.V.; Simmons, D.L. The Cyclooxygenases. Genome Biol. 2004. 24, 1-7.
[13] Snipes, K.B.; Busija, J.A. Acetaminophen and the Cyclooxygenase-3
Puzzle: Sorting out Facts, Fictions, and Uncertainties. JPET. 2005, 315, 1-7.
[14] Hinz, B.; Cheremina, O.; Brune. K. Acetaminophen (paracetamol) is a
selective
cyclooxygenase-2 inhibitor in man. FASEB J. 2008,
22, 383-390.
[15] Dannhardt, G.; Kiefer, W. Cyclooxygenase Inhibitors-Current Status and
Future Prospects. Eur. J. Med. Chem. 2001, 36, 109-126.
[16] Rang H.P; Ritter, J.M.; Dale M.M. Pharmacology. 5th Ed.
Churchill Livingstone, London, UK, 2003, pp. 247-252.
[17]
Laine
, L.;
Takeuchi
, K.;
Tarnawski
, A. Gastric Mucosal Defense and Cytoprotection: Bench to Bedside. Gastroenterology. 2008,
135
,
41-60.
[18]
Vane, J.R.;
Botting
, R. M. Mechanism of Action of Nonsteroidal Anti-inflammatory Drugs,
The American Journal of Medicine
. 1998,
104,
2–8.
[19] Kalgutkar, A.S.; Zhiyang, Z. Discovery and Design of Selective
Cyclooxygenase-2
Inhibitors as Non-Ulcerogenic, Anti-Inflammatory Drugs with Potential
Utility as Anti-
Cancer Agents. Current Drug Targe., 2001, 2, 79-106.
[20] Fiorucci, S.; Rosaria, M.; Bucci, C.G. Dual
Inhibitors of Cyclooxygenase and 5-
Lipooxygenase. A New Avenue in Anti-Inflammatory Therapy?
Biochem. Pharmacol.
2001
, 62,1433-1438.
[21]
Gaur
, K.;
Kori
, M.L.;
Tyagi
, L.K.;
Singh
, V.;
Nema
, R.K.;
Tripathi
, P.;
Sharma
, C.S. Licofelone- novel analgesic and anti-inflammatory agent for
osteoarthritis: An overview. J Young Pharmacists. 2009, 1, 67-71.
[
22]
Pontiki
, E.;
Litina
, D.H.;
Litinas
, K.;
Nicolotti
, O.;
Carotti
, A. Design, synthesis and pharmacobiological evaluation of novel acrylic
acid derivatives acting as lipoxygenase and cyclooxygenase-1 inhibitors
with antioxidant and anti-inflammatory activities.
European Journal of Medicinal Chemistry
. 2011,
46
,
191–200.
[23] Velazquez, C; Rao, P.N.P.; Knaus, E.E. Novel Nonsteroidal
Antiinflammatory Drugs Possessing a Nitric Oxide DonorDiazen-1
-ium-1,2-diolate Moiety: Design, Synthesis, Biological Evaluation, and
Nitric Oxide Release Studies. J. Med. Chem. 2005, 48, 4061-
4067.
[24]
Kashfi
, K.;
Olson
, K.R. Biology and therapeutic potential of hydrogen sulfide and hydrogen
sulfide-releasing chimeras.
Biochemical Pharmacology
.
2012
,
In Press,
[25]
http://www.topnews.in/rotten-eggs-may-pave-way-safer-antiinflammatory-drugs-2169214.
(Visited, February 2013)